

Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation
María M. Caffarel, David Sarrió, José Palacios, Manuel Guzmán and Cristina Sánchez
DOI: 10.1158/0008-5472.CAN-05-4566 Published July 2006
Abstract
It has been proposed that cannabinoids are involved in the control of cell fate. Thus, these compounds can modulate proliferation, differentiation, and survival in different manners depending on the cell type and its physiopathologic context. However, little is known about the effect of cannabinoids on the cell cycle, the main process controlling cell fate. Here, we show that Δ9-tetrahydrocannabinol (THC), through activation of CB2 cannabinoid receptors, reduces human breast cancer cell proliferation by blocking the progression of the cell cycle and by inducing apoptosis. In particular, THC arrests cells in G2-M via down-regulation of Cdc2, as suggested by the decreased sensitivity to THC acquired by Cdc2-overexpressing cells. Of interest, the proliferation pattern of normal human mammary epithelial cells was much less affected by THC. We also analyzed by real-time quantitative PCR the expression of CB1 and CB2 cannabinoid receptors in a series of human breast tumor and nontumor samples. We found a correlation between CB2expression and histologic grade of the tumors. There was also an association between CB2expression and other markers of prognostic and predictive value, such as estrogen receptor, progesterone receptor, and ERBB2/HER-2 oncogene. Importantly, no significant CB2 expression was detected in nontumor breast tissue. Taken together, these data might set the bases for a cannabinoid therapy for the management of breast cancer.(Cancer Res 2006; 66(13): 6615-21)
Introduction
There are very few critical decisions that cells must take during their lifetime. Basically, these are whether to proliferate, differentiate, or die. A tight regulation of the cell cycle is crucial to control all these decisions, and its deregulation has devastating consequences, such as cancer ( 1). It has been proposed that cannabinoids, the active components of Cannabis sativa, play a role in the control of the aforementioned decisions. For example, they can modulate survival, proliferation, and differentiation depending on the cell type and its physiopathologic context ( 2, 3). Among the ∼70 cannabinoids synthesized by C. sativa, Δ9-tetrahydrocannabinol (THC) is the most important in terms of potency and abundance ( 4). THC exerts a wide variety of biological effects by mimicking endogenous compounds, the endocannabinoids anandamide and 2-arachidonoylglycerol, which activate specific cannabinoid receptors. Thus far, two G protein–coupled cannabinoid-specific receptors have been cloned from mammalian tissues: CB1, abundantly expressed in the brain and at many peripheral sites, and CB2, almost exclusively expressed in the immune system ( 5). Engagement of these receptors by THC or endocannabinoids affects several signaling pathways, some of them directly involved in the control of cell fate. For instance, cannabinoids modulate mitogen-activated protein kinases and the phosphatidylinositol 3-kinase/Akt survival pathway, which have a prominent role in the control of cell growth and differentiation ( 6). Due to the growing evidence that cannabinoids participate in the control of cell fate and to the fact that the cell cycle is a key process underlying the regulation of survival/proliferation/differentiation decisions, we decided to study the effect of THC on the cell cycle and the mechanism of cannabinoid action on this process. Because breast tumors are one of the most common human neoplasias and one of the leading causes of death among Western women ( 7), we decided to focus our studies on this particular type of cancer.
Materials and Methods
Cell culture and viability. EVSA-T, MDA-MB-231, MDA-MB-468, and SKBr3 cells were kindly given by Dr. López-Rivas [Centro Andaluz de Biología del Desarrollo, Consejo Superior de Investigaciones Científicas (CSIC), Sevilla, Spain], and MCF-7 and T-47D cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Cells were maintained in RPMI 1640 (EVSA-T, MDA-MB-231, MCF-7, and T-47D) or DMEM (MDA-MB-468 and SKBr3) supplemented with 10% fetal bovine serum (FBS), 5 units/mL penicillin, and 5 mg/mL streptomycin. Human mammary epithelial cells (HMEC) were kindly given by Dr. Lacal (Instituto de Investigaciones Biomédicas, CSIC, Madrid, Spain) and grown in mammary epithelial growth medium (Cambrex, East Rutherford, NJ) according to the manufacturer's instructions. Cannabinoid ligands were prepared in DMSO. Control incubations had the corresponding DMSO content. No significant influence of DMSO was observed on cell viability at the final concentration used (0.1-0.2%, v/v). Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide test (Sigma, St. Louis, MO) according to the manufacturer's instructions.
Western blot analysis. Samples were subjected to SDS-PAGE, and proteins were transferred onto polyvinylidene fluoride membranes. Blots were incubated with the following antibodies: anti-phosphorylated Cdc2 (Tyr15), anti-cyclin B1, anti-Cdc25C, anti-p27, anti-caspase-3, and anti-poly(ADP-ribose) polymerase (PARP) (Cell Signaling, Beverly, MA); anti-Cdc2 and anti-p21 (Santa Cruz, Santa Cruz, CA); anti-Wee1 (BioVision, Mountain View, CA); anti-survivin (R&D Systems, Minneapolis, MN); and anti-α-tubulin as loading control (Sigma). Luminograms were obtained with an enhanced chemiluminescence detection kit (Amersham Life Sciences, Arlington Heights, IL), and densitometric analysis was done with Multianalyst software (Bio-Rad, Hercules, CA).
Cell cycle analysis. Cells were permeabilized and fixed in 1% (w/v) bovine serum albumin and 30% ethanol-PBS and labeled with 5 μg/mL Hoechst 33342 (Molecular Probes, Leiden, the Netherlands). Fluorescence intensity was analyzed using a LSR flow cytometer (Becton Dickinson, San Jose, CA). Ten thousand cells per analysis were recorded.
Apoptosis. Cells were incubated in binding buffer [10 mmol/L HEPES (pH 7.4), 2.5 mmol/L CaCl2, 140 mmol/L NaCl] supplemented with Annexin V-FITC (Molecular Probes). Propidium iodide (PI; Sigma) was added 1 minute before sample analysis. Fluorescence intensity was analyzed using a FACS Scalibur flow cytometer (Becton Dickinson). For triple staining experiments, cells were labeled with Hoechst 33342, and apoptosis was analyzed as described above. Ten thousand cells per analysis were recorded.
Caspase-3 activity. Caspase-3/7 activity was determined with a luminogenic substrate (Caspase-Glo, Promega, Madison, WI) according to the manufacturer's instructions. Luminescence was determined in a Microplate Fluorescence Reader (BMG Labtech, Offenburg, Germany).
Tissue samples. Samples were obtained from the Centro Nacional de Investigaciones Oncológicas Tumor Bank (Madrid, Spain). Histologic grade was assessed according to Elston and Ellis criteria. Immunohistochemical staining for prognostic and predictive factors was done by the EnVision method with a heat-induced antigen retrieval step. Monoclonal antibodies for estrogen receptor (ER), progesterone receptor (PR), p53 (Novocastra, Newcastle, United Kingdom), and Ki67 (DAKO, Glostrup, Denmark) were used. ERBB2/HER-2 expression was evaluated using Herceptest (DAKO). The percentage of cells with unequivocal nuclear staining for ER, PR, Ki67, and p53 was scored, and a cutoff of 5% was used for positivity for ER and PR and 15% for Ki67 and p53. For ERBB2/HER-2, only cases with 3+ membranous staining were scored as positive.
Confocal microscopy analysis of cannabinoid receptors. Human breast cancer and normal breast 5-μm paraffin-embedded tissue sections were analyzed. Primary antibodies against CB1 and CB2 receptors (Affinity Bioreagents, Golden, CO) were used. Secondary anti-rabbit antibody Alexa Fluor 594 was from Molecular Probes. Cell nuclei were stained with YOYO-1 (Molecular Probes). Confocal fluorescence images were acquired using Laser Sharp 2000 software (Bio-Rad).
Reverse transcription-PCR analysis. Total RNA was isolated using the RNeasy Protect kit (Qiagen, Hilden, Germany). cDNA was obtained using Transcriptor Reverse Transcriptase (Roche, Applied Science, Penzberg, Germany). Primer sequences were CB1 (sense), 5′-CGTGGGCAGCCTGTTCCTCA-3′; CB1 (antisense), 5′-CATGCGGGCTTGGTCTGG-3′; CB2(sense), 5′-TGGGACAGGGTCAGTACAAGT-3′; CB2 (antisense), 5′-CTTTGGCTCCTGGTGGTCT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH; sense), 5′-GGGAAGCTCACTGGCATGGCCTTCC-3′; and GAPDH (antisense), 5′-CATGTGGGCCATGAGGTCCACCAC-3′.
Real-time quantitative PCR. Total RNA and cDNA were obtained from frozen breast tumors or cell lines as described above. Taqman probes for human CB1, CB2, and 18S RNA (used as internal reference) were from Applied Biosystems (Foster City, CA).
Plasmids and transfections. pIRESpuro2 and pIRESpuro2-Cdc2 were kindly given by Dr. Huang (Johns Hopkins University, Baltimore, MD). Transfections were carried out with Fugene 6 (Roche Applied Science, Indianapolis, IN) according to the manufacturer's protocol. Transfected cells were selected with puromycin.
Statistical analysis. ANOVA with a post hoc analysis by the Student's-Newman-Keuls' test was routinely used. For cannabinoid receptor expression in human samples, data were log transformed to achieve normality in the distribution. An F-test was subsequently done to compare equality of variances in each group, and a classic t test or a t test with different variances was applied using the Welch modification.
Results
THC inhibits proliferation of human breast cancer cells. Several human breast cell lines were incubated with THC, and viable cell numbers were estimated. THC decreased proliferation in all the tumor cells tested ( Fig. 1A ). Among the tumor cells, those with more aggressive phenotype (ER−) were more sensitive to THC ( Fig. 1B). Remarkably, nontumor HMEC cells were the most resistant to cannabinoid treatment (IC50 >12 μmol/L; Fig. 1A). Rimonabant, a selective CB1 receptor antagonist, did not block THC effect in EVSA-T cells ( Fig. 1C) or any of the other cell lines studied (data not shown). In contrast, SR144528, a selective CB2 receptor antagonist, partially prevented the THC-induced decrease of EVSA-T cell proliferation ( Fig. 1C). Both reverse transcription-PCR (RT-PCR; Fig. 1D) and real-time quantitative PCR experiments ( Fig. 1B) confirmed the expression of CB2 mRNA in this cell line, whereas CB1 mRNA was undetectable ( Fig. 1B and D).
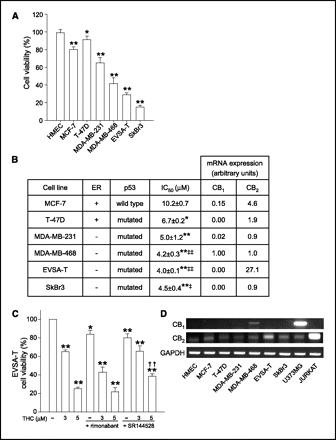
Figure 1.
Effect of cannabinoids on human breast cell proliferation. Cells were incubated in low-FBS medium (2.5% for MCF-7 and 0.5% for the rest of the tumor cell lines) for 72 hours in the presence of 5 μmol/L THC (A), different concentrations of THC ranging from 1 to 12 μmol/L (B), or the specified THC concentration (C). When antagonists were used (1 μmol/L rimonabant and 2 μmol/L SR144528), they were added 1 hour before THC. ER and p53 status was obtained from ATCC and German National Resource Centre for Biological Material (DSMZ, Braunschweig, Germany). IC50s were defined as concentrations of THC required to decrease cell viability to 50%. A, columns, mean for each cell line (n ≥ 3); bars, SE. B, significant differences from MCF-7 (*, P < 0.05; **, P < 0.01) or T-47D cells (‡, P < 0.05; ‡‡, P < 0.01). For real-time quantitative PCR experiments, an arbitrary value of 1 was assigned to cannabinoid receptor expression in MDA-MB 468 cells (reference). Representative experiment (n = 3). A and C, columns, means cell viability in THC-treated cells versus their respective vehicle-treated cells (set at 100%; n ≥ 3); bars, SE. Significant differences (*, P < 0.05; **, ††, P < 0.01) from control (*) or 5 μmol/L THC alone (†). D, CB1 and CB2 mRNA expression in the cell lines was determined by RT-PCR (representative gels, n ≥ 3). U373 MG astrocytoma cells and Jurkat leukemia cells were used as positive controls for CB1 and CB2, respectively.
THC-induced decrease of cell proliferation is due to the blockade of the G2-M transition. We next sought to examine whether an alteration of EVSA-T cell cycle underlies THC antiproliferative effect. The cannabinoid increased the number of cells in the G0-G1 compartment and, in parallel, decreased the number of cells in S phase ( Fig. 2A and C ). At the highest concentration tested (5 μmol/L), THC also produced the following: (a) an increase in the number of cells in G2-M phases and (b) the appearance of a population of hypodiploid cells ( Fig. 2A and C). The latter two effects were prevented by SR144528 ( Fig. 2C). Importantly, THC did not alter the cell cycle profile of HMEC cells ( Fig. 2B).
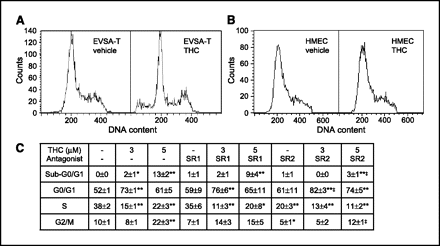
Figure 2.
Effect of THC on cell cycle dynamics. Cell cycle profiles of EVSA-T (A) and HMEC cells (B) incubated with 5 μmol/L THC or the corresponding vehicle for 48 hours. Representative experiments (n ≥ 3). C, percentage of EVSA-T cells (mean ± SE) in every phase of the cell cycle after 48 hours of incubation with the indicated compounds. Where indicated, rimonabant (SR1; 1 μmol/L) or SR144528 (SR2; 2 μmol/L) was added 1 hour before THC. Significant differences from control (*, P < 0.05; **, P < 0.01) or the corresponding concentration of THC alone (‡, P < 0.05).
To analyze the precise mechanism of THC action, we first studied the expression of several proteins involved in the G2-M transition. THC decreased the total levels of Cdc2 [p34, cyclin-dependent kinase (CDK) 1; Fig. 3A ], the major CDK controlling the entrance of cells in mitosis after completing G2 events ( 8), and SR144528 (2 μmol/L) completely prevented this effect [relative optical density (OD) after 16 hours of treatment relative to vehicle: SR144528+THC, 105 ± 4]. The expression of cyclin B1, the positive regulatory subunit of Cdc2 ( 9), did not significantly change on cannabinoid challenge (data not shown). The levels of p21, a CDK inhibitor known to prevent Cdc2-cyclin B activation ( 8), were enhanced by THC ( Fig. 3A). It has been recently proposed that p27, a CDK inhibitor traditionally associated to the regulation of G1-S transition, can also inhibit Cdc2 at G2-M (10). In our system, however, THC did not modify p27 levels (data not shown).
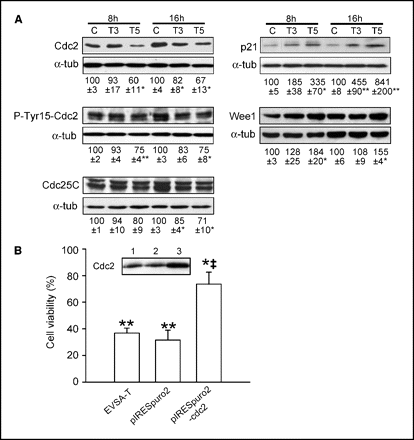
Figure 3.
Effect of THC on the expression of proteins involved in the control of the G2-M transition. A, Western blot analysis of the indicated proteins was done at least twice. Representative luminograms. T3, 3 μmol/L THC and T5, 5 μmol/L THC. Optical density (arbitrary units, mean ± SE) of the different proteins relative to their respective time-point control incubations (set at 100). B, EVSA-T cell viability was assayed after 72 hours of incubation with 5 μmol/L THC or vehicle. Columns, mean cell viability in THC-treated cells versus their respective vehicle-treated cells (set at 100%); bars, SE. Inset, Western blot of total Cdc2 levels in EVSA-T cells (lane 1), EVSA-T cells transfected with pIRESpuro2 (lane 2), and EVSA-T cells transfected with pIRESpuro2-Cdc2 (lane 3). A and B, significant differences from the corresponding vehicle-treated cells (*, P < 0.05; **, P < 0.01) or between THC-treated cells transfected with either pIRESpuro2 or pIRESpuro2-Cdc2 (‡, P < 0.05).
To be active, Cdc2 has to be dephosphorylated in the Tyr15 residue ( 11). THC treatment decreased the amount of phosphorylated Tyr15-Cdc2 to a lower extent than total Cdc2 levels ( Fig. 3A), indicating that the ratio inactive/active Cdc2 was augmented by THC. Phosphorylation of Cdc2 in Tyr15 is controlled by the Wee1/Mik1 family of protein kinases and by the phosphatase Cdc25C (11). Our results show that THC enhances Wee1 and reduces Cdc25C protein levels ( Fig. 3A).
In view of the aforementioned results, it is conceivable that THC exposure prevents EVSA-T cells to reach the required levels of active Cdc2 to enter mitosis. To test whether Cdc2 down-regulation is important in the growth-inhibiting effect of THC, we heterologously expressed this kinase. As shown in Fig. 3B, cells overexpressing Cdc2 became significantly more resistant to THC.
THC-induced cell cycle arrest is associated with apoptosis. We next tried to elucidate whether THC-induced inhibition of proliferation was associated with cell death. The cannabinoid induced apoptosis, a process that was prevented by SR144528 ( Fig. 4A ). THC also induced a two-fold increase in caspase-3 activity, an effect that was prevented by SR144528 ( Fig. 4B). Likewise, we observed reduced levels of both pro-caspase-3 (the inactive precursor of caspase-3) and PARP (a caspase-3 substrate) in cannabinoid-treated cells ( Fig. 4C). We subsequently addressed the question of whether apoptotic cells were those arrested in G2-M by THC. We conducted triple staining experiments to analyze the percentage of apoptotic cells in every phase of the cell cycle. As shown in Fig. 5A , THC induced apoptosis in all the cell cycle phases, but the majority of apoptotic cells were in the G2-M compartment.
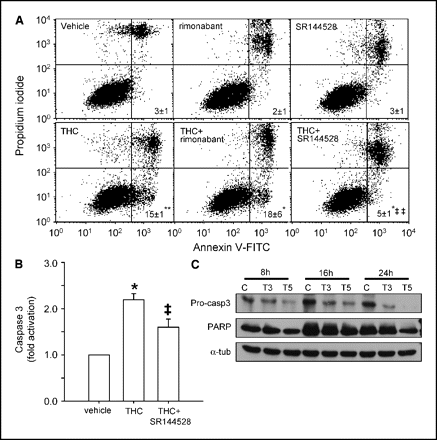
Figure 4.
Analysis of THC-induced cell death. Fluorescence-activated cell sorting analysis (A) and caspase-3 activity (B) of EVSA-T cells after incubation with the indicated compounds for 24 hours. Antagonists (1 μmol/L rimonabant or 2 μmol/L SR144528) were added 1 hour before THC (5 μmol/L). A, percentage of apoptotic cells (FITC positive/PI negative) within the total cell population. B, columns, means of at least five or more experiments; bars, SE. Significant differences from control (*, P < 0.05; **, P < 0.01) or THC alone (‡, P < 0.05; ‡‡, P < 0.01). C, Western blot analysis of pro-caspase-3 and PARP. T3, 3 μmol/L THC and T5, 5 μmol/L THC. Representative luminograms (n ≥ 3).
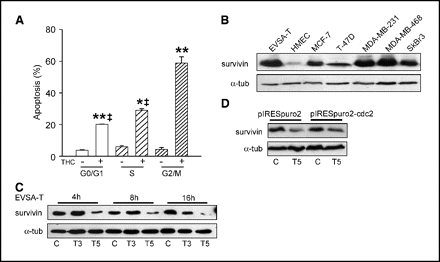
Figure 5.
Connection between cell cycle arrest and apoptosis. A, percentage of apoptotic cells in different phases of the cell cycle as assessed by triple staining with Annexin V-FITC, PI, and Hoechst 33342. Significant differences from the corresponding vehicle-treated cells (*, P < 0.05; **, P < 0.01) or between THC-treated cells in G2-M and THC-treated cells in G0-G1 or S (‡, P< 0.05). B-D, Western blot of survivin in different human breast cell lines (B), EVSA-T cells incubated with THC (T3, 3 μmol/L THC; T5, 5 μmol/L THC) or the corresponding vehicle (C), and EVSA-T cells transfected with pIRESpuro2 or pIRESpuro-Cdc2 and incubated for 16 hours with the indicated compounds (D). Representative luminograms (n ≥ 2).
It is known that survivin, a member of the inhibitor of apoptosis family, can be phosphorylated in Thr34 by Cdc2. This phosphorylation results in enhanced stability of survivin and the consequent inhibition of caspase activity ( 12). Survivin was highly expressed in all the breast cancer cell lines tested but was hardly detectable in HMEC ( Fig. 5B), in agreement with previous reports showing a sharp differential expression in cancer (high levels) versus normal (undetectable levels) tissues (12). Moreover, THC decreased survivin levels in EVSA-T cells ( Fig. 5C), which may explain why THC-induced Cdc2 inactivation results in apoptosis. In fact, when Cdc2 levels were enhanced by overexpression, survivin decrease on cannabinoid treatment was attenuated (relative optical density after 16 hours of treatment for pIRESpuro2-transfected cells: vehicle, 100 ± 6; 5 μmol/L THC, 31 ± 4. For pIRESpuro2-Cdc2-transfected cells: vehicle, 100 ± 5; 5 μmol/L THC, 74 ± 3; Fig. 5D).
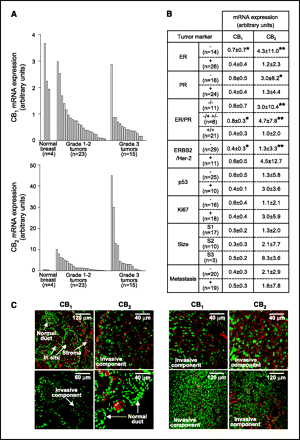
Cannabinoid receptors are expressed in human breast tumors. The presence of CB1 and CB2receptors in human breast tumors was evaluated by real-time quantitative PCR and confocal microscopy. Lower levels of CB1 mRNA were detected in tumors of low-medium (grade 1-2) and high (grade 3) histologic grade compared with normal, noncancerous breast tissue (grade 1-2 versus noncancerous breast tissue (P = 0.008); grade 3 versus noncancerous breast tissue (P = 0.0007); Fig. 6A ). CB2 expression was higher than CB1 expression in all the tumors analyzed (P = 0.00002) and seemed to correlate with their histologic grade (grade 1-2 versus grade 3; P = 0.04; Fig. 6A). Of interest, CB2 transcripts were hardly detectable in normal breast tissue ( Fig. 6A). Moreover, the expression of CB2 showed an association with molecular markers of prognostic value. Thus, ER− tumors expressed more CB2 mRNA than ER+ tumors ( Fig. 6B). CB2 expression was also higher in PR− than in PR+ samples. ER−/PR− tumors (response rate <10% to conventional therapies) expressed more CB2 mRNA than ER+/PR+ tumors (60-70% response rate; ref. 7; Fig. 6B). ERBB2/HER-2-positive tumors (with worse survival prediction at least in node-positive patients; ref. 7) expressed higher levels of CB2 mRNA than ERBB2/HER-2-negative tumors ( Fig. 6B). Confocal microscopy experiments confirmed the presence of CB1 and CB2 proteins in tumor cells ( Fig. 6C). None of the receptor proteins was detected in normal ducts.
There is scant information on the effect of cannabinoids on the cell cycle. It has been shown previously that the endogenous cannabinoid anandamide arrests the cycle of hepatoma HepG2 cells ( 13), epidermal growth factor–stimulated PC3 prostate cancer cells ( 14), and the breast cancer cell line EFM-19 ( 15) at the G1-S transition. The same effect was exerted by a metabolically stable analogue of anandamide, met-F-anandamide, on KiMol thyroid carcinoma cells ( 16). Results presented herein (a) show that the plant-derived cannabinoid THC is able to block the progression of breast cancer cell cycle and (b) provide a mechanism for this action. Of interest, nontumor mammary epithelial cells were rather insensitive to THC, suggesting that cannabinoids would fulfill one of the demanded requirements of any compound intended to be used in cancer therapy: the selectivity for tumor cells. In our experimental model, THC induces a CB2-mediated cell cycle arrest at the G2-M transition via Cdc2 down-regulation. We are nonetheless aware that our observation that THC effects are significantly but not completely prevented by the CB2 selective antagonist also points to the existence of CB2-independent processes in cannabinoid antiproliferative action. Most cancer cells evade antigrowth signals because they have defective G1 checkpoints, which makes the G2 checkpoint an attractive target for cancer therapy ( 11). Since its discovery in 1986 ( 17), it has been clear that Cdc2 is essential for cell cycle progression ( 18). In fact, Cdc2-deficient mice die at very early stages of embryonic development ( 18). The pivotal importance of this particular protein could explain why, in our system, a somewhat moderate decrease of total Cdc2 levels (∼40%) results in large changes in cell viability. In addition, our data indicate that THC increases the inactive/active Cdc2 ratio, supporting that the cannabinoid decreases not only total Cdc2 levels but also enzyme-specific activity.
It is important to point out that, although the antiproliferative effect of cannabinoids on different tumor cells has been extensively confirmed both in vitro and in vivo ( 6), a recent report indicates that THC may enhance breast cancer cell growth under certain circumstances. In that study, the authors showed a direct association between the degree of sensitivity of a tumor to THC and the level of CB1 and CB2 expression. Thus, THC has antiproliferative effect in tumors expressing cannabinoid receptors, whereas those with low to no expression suffer increased growth and metastasis due to THC-induced suppression of the antitumor immune response ( 19). Results presented herein are not in disagreement with that report, as EVSA-T cells, which are very sensitive to THC, express high levels of CB2.
By real-time quantitative PCR experiments, we have observed a correlation between CB2expression and the histologic grade of breast tumors. Moreover, CB2 expression is higher in tumors with predicted low response to conventional therapies, for instance ER−/PR− tumors, which are weakly responsive to adjuvant tamoxifen ( 7). In addition, hardly detectable levels of CB2 were found in normal breast tissue. Because both cell cycle arrest and apoptosis induced by THC are CB2-mediated effects, it is tempting to speculate that the tumors with poor prognosis (i.e., those reluctant to conventional therapies and, according to our data, expressing the highest levels of CB2receptor), may be the most responsive to cannabinoids. In addition, the psychotropic effects of cannabinoids are mediated by CB1 but not CB2 receptors, and, therefore, a cannabinoid-based therapy selectively targeting CB2 receptors would be devoid of the side effects associated to cannabis consumption ( 20).
Breast cancer is the most common malignant disease among Western women. Although the rates of mortality of breast cancer patients have decreased as a result of early diagnosis by mammograms, certain breast tumors remain reluctant to conventional therapies, and current treatments have side effects that substantially affect the patient's quality of life ( 21). Our findings might set the basis for new strategies for the management of breast cancer.
Acknowledgments
Grant support: Fondo de Investigaciones Sanitarias FIS 04/1029 (C. Sánchez) and BEFI 01/9132 (D. Sarrió); Complutense University PR1/05 (C. Sánchez); Ministerio de Educación y Ciencia SAF2003-00745 (M. Guzmán), SAF2004-08258-C02-01 (J. Palacios), and AP2003-0063 (M.M. Caffarel); and Fundación Científica de la Asociación Española Contra el Cáncer (M. Guzmán).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank the staff at Centro de Microscopía y Citometría (Complutense University, Madrid, Spain) and A. Álvarez-Barrientos for expert advice on flow cytometry experiments; O. Rueda for expert advice on statistical analysis; and the members of our laboratory for technical support, especially to K. Blazej, C. Blázquez, D. Callegarin, A. Egia, B. Herrera, and M. Lorente.
Footnotes
María M. Caffarel, David Sarrió, José Palacios, Manuel Guzmán and Cristina Sánchez
DOI: 10.1158/0008-5472.CAN-05-4566 Published July 2006
Abstract
It has been proposed that cannabinoids are involved in the control of cell fate. Thus, these compounds can modulate proliferation, differentiation, and survival in different manners depending on the cell type and its physiopathologic context. However, little is known about the effect of cannabinoids on the cell cycle, the main process controlling cell fate. Here, we show that Δ9-tetrahydrocannabinol (THC), through activation of CB2 cannabinoid receptors, reduces human breast cancer cell proliferation by blocking the progression of the cell cycle and by inducing apoptosis. In particular, THC arrests cells in G2-M via down-regulation of Cdc2, as suggested by the decreased sensitivity to THC acquired by Cdc2-overexpressing cells. Of interest, the proliferation pattern of normal human mammary epithelial cells was much less affected by THC. We also analyzed by real-time quantitative PCR the expression of CB1 and CB2 cannabinoid receptors in a series of human breast tumor and nontumor samples. We found a correlation between CB2expression and histologic grade of the tumors. There was also an association between CB2expression and other markers of prognostic and predictive value, such as estrogen receptor, progesterone receptor, and ERBB2/HER-2 oncogene. Importantly, no significant CB2 expression was detected in nontumor breast tissue. Taken together, these data might set the bases for a cannabinoid therapy for the management of breast cancer.(Cancer Res 2006; 66(13): 6615-21)
Introduction
There are very few critical decisions that cells must take during their lifetime. Basically, these are whether to proliferate, differentiate, or die. A tight regulation of the cell cycle is crucial to control all these decisions, and its deregulation has devastating consequences, such as cancer ( 1). It has been proposed that cannabinoids, the active components of Cannabis sativa, play a role in the control of the aforementioned decisions. For example, they can modulate survival, proliferation, and differentiation depending on the cell type and its physiopathologic context ( 2, 3). Among the ∼70 cannabinoids synthesized by C. sativa, Δ9-tetrahydrocannabinol (THC) is the most important in terms of potency and abundance ( 4). THC exerts a wide variety of biological effects by mimicking endogenous compounds, the endocannabinoids anandamide and 2-arachidonoylglycerol, which activate specific cannabinoid receptors. Thus far, two G protein–coupled cannabinoid-specific receptors have been cloned from mammalian tissues: CB1, abundantly expressed in the brain and at many peripheral sites, and CB2, almost exclusively expressed in the immune system ( 5). Engagement of these receptors by THC or endocannabinoids affects several signaling pathways, some of them directly involved in the control of cell fate. For instance, cannabinoids modulate mitogen-activated protein kinases and the phosphatidylinositol 3-kinase/Akt survival pathway, which have a prominent role in the control of cell growth and differentiation ( 6). Due to the growing evidence that cannabinoids participate in the control of cell fate and to the fact that the cell cycle is a key process underlying the regulation of survival/proliferation/differentiation decisions, we decided to study the effect of THC on the cell cycle and the mechanism of cannabinoid action on this process. Because breast tumors are one of the most common human neoplasias and one of the leading causes of death among Western women ( 7), we decided to focus our studies on this particular type of cancer.
Materials and Methods
Cell culture and viability. EVSA-T, MDA-MB-231, MDA-MB-468, and SKBr3 cells were kindly given by Dr. López-Rivas [Centro Andaluz de Biología del Desarrollo, Consejo Superior de Investigaciones Científicas (CSIC), Sevilla, Spain], and MCF-7 and T-47D cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Cells were maintained in RPMI 1640 (EVSA-T, MDA-MB-231, MCF-7, and T-47D) or DMEM (MDA-MB-468 and SKBr3) supplemented with 10% fetal bovine serum (FBS), 5 units/mL penicillin, and 5 mg/mL streptomycin. Human mammary epithelial cells (HMEC) were kindly given by Dr. Lacal (Instituto de Investigaciones Biomédicas, CSIC, Madrid, Spain) and grown in mammary epithelial growth medium (Cambrex, East Rutherford, NJ) according to the manufacturer's instructions. Cannabinoid ligands were prepared in DMSO. Control incubations had the corresponding DMSO content. No significant influence of DMSO was observed on cell viability at the final concentration used (0.1-0.2%, v/v). Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide test (Sigma, St. Louis, MO) according to the manufacturer's instructions.
Western blot analysis. Samples were subjected to SDS-PAGE, and proteins were transferred onto polyvinylidene fluoride membranes. Blots were incubated with the following antibodies: anti-phosphorylated Cdc2 (Tyr15), anti-cyclin B1, anti-Cdc25C, anti-p27, anti-caspase-3, and anti-poly(ADP-ribose) polymerase (PARP) (Cell Signaling, Beverly, MA); anti-Cdc2 and anti-p21 (Santa Cruz, Santa Cruz, CA); anti-Wee1 (BioVision, Mountain View, CA); anti-survivin (R&D Systems, Minneapolis, MN); and anti-α-tubulin as loading control (Sigma). Luminograms were obtained with an enhanced chemiluminescence detection kit (Amersham Life Sciences, Arlington Heights, IL), and densitometric analysis was done with Multianalyst software (Bio-Rad, Hercules, CA).
Cell cycle analysis. Cells were permeabilized and fixed in 1% (w/v) bovine serum albumin and 30% ethanol-PBS and labeled with 5 μg/mL Hoechst 33342 (Molecular Probes, Leiden, the Netherlands). Fluorescence intensity was analyzed using a LSR flow cytometer (Becton Dickinson, San Jose, CA). Ten thousand cells per analysis were recorded.
Apoptosis. Cells were incubated in binding buffer [10 mmol/L HEPES (pH 7.4), 2.5 mmol/L CaCl2, 140 mmol/L NaCl] supplemented with Annexin V-FITC (Molecular Probes). Propidium iodide (PI; Sigma) was added 1 minute before sample analysis. Fluorescence intensity was analyzed using a FACS Scalibur flow cytometer (Becton Dickinson). For triple staining experiments, cells were labeled with Hoechst 33342, and apoptosis was analyzed as described above. Ten thousand cells per analysis were recorded.
Caspase-3 activity. Caspase-3/7 activity was determined with a luminogenic substrate (Caspase-Glo, Promega, Madison, WI) according to the manufacturer's instructions. Luminescence was determined in a Microplate Fluorescence Reader (BMG Labtech, Offenburg, Germany).
Tissue samples. Samples were obtained from the Centro Nacional de Investigaciones Oncológicas Tumor Bank (Madrid, Spain). Histologic grade was assessed according to Elston and Ellis criteria. Immunohistochemical staining for prognostic and predictive factors was done by the EnVision method with a heat-induced antigen retrieval step. Monoclonal antibodies for estrogen receptor (ER), progesterone receptor (PR), p53 (Novocastra, Newcastle, United Kingdom), and Ki67 (DAKO, Glostrup, Denmark) were used. ERBB2/HER-2 expression was evaluated using Herceptest (DAKO). The percentage of cells with unequivocal nuclear staining for ER, PR, Ki67, and p53 was scored, and a cutoff of 5% was used for positivity for ER and PR and 15% for Ki67 and p53. For ERBB2/HER-2, only cases with 3+ membranous staining were scored as positive.
Confocal microscopy analysis of cannabinoid receptors. Human breast cancer and normal breast 5-μm paraffin-embedded tissue sections were analyzed. Primary antibodies against CB1 and CB2 receptors (Affinity Bioreagents, Golden, CO) were used. Secondary anti-rabbit antibody Alexa Fluor 594 was from Molecular Probes. Cell nuclei were stained with YOYO-1 (Molecular Probes). Confocal fluorescence images were acquired using Laser Sharp 2000 software (Bio-Rad).
Reverse transcription-PCR analysis. Total RNA was isolated using the RNeasy Protect kit (Qiagen, Hilden, Germany). cDNA was obtained using Transcriptor Reverse Transcriptase (Roche, Applied Science, Penzberg, Germany). Primer sequences were CB1 (sense), 5′-CGTGGGCAGCCTGTTCCTCA-3′; CB1 (antisense), 5′-CATGCGGGCTTGGTCTGG-3′; CB2(sense), 5′-TGGGACAGGGTCAGTACAAGT-3′; CB2 (antisense), 5′-CTTTGGCTCCTGGTGGTCT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH; sense), 5′-GGGAAGCTCACTGGCATGGCCTTCC-3′; and GAPDH (antisense), 5′-CATGTGGGCCATGAGGTCCACCAC-3′.
Real-time quantitative PCR. Total RNA and cDNA were obtained from frozen breast tumors or cell lines as described above. Taqman probes for human CB1, CB2, and 18S RNA (used as internal reference) were from Applied Biosystems (Foster City, CA).
Plasmids and transfections. pIRESpuro2 and pIRESpuro2-Cdc2 were kindly given by Dr. Huang (Johns Hopkins University, Baltimore, MD). Transfections were carried out with Fugene 6 (Roche Applied Science, Indianapolis, IN) according to the manufacturer's protocol. Transfected cells were selected with puromycin.
Statistical analysis. ANOVA with a post hoc analysis by the Student's-Newman-Keuls' test was routinely used. For cannabinoid receptor expression in human samples, data were log transformed to achieve normality in the distribution. An F-test was subsequently done to compare equality of variances in each group, and a classic t test or a t test with different variances was applied using the Welch modification.
Results
THC inhibits proliferation of human breast cancer cells. Several human breast cell lines were incubated with THC, and viable cell numbers were estimated. THC decreased proliferation in all the tumor cells tested ( Fig. 1A ). Among the tumor cells, those with more aggressive phenotype (ER−) were more sensitive to THC ( Fig. 1B). Remarkably, nontumor HMEC cells were the most resistant to cannabinoid treatment (IC50 >12 μmol/L; Fig. 1A). Rimonabant, a selective CB1 receptor antagonist, did not block THC effect in EVSA-T cells ( Fig. 1C) or any of the other cell lines studied (data not shown). In contrast, SR144528, a selective CB2 receptor antagonist, partially prevented the THC-induced decrease of EVSA-T cell proliferation ( Fig. 1C). Both reverse transcription-PCR (RT-PCR; Fig. 1D) and real-time quantitative PCR experiments ( Fig. 1B) confirmed the expression of CB2 mRNA in this cell line, whereas CB1 mRNA was undetectable ( Fig. 1B and D).
Figure 1.
Effect of cannabinoids on human breast cell proliferation. Cells were incubated in low-FBS medium (2.5% for MCF-7 and 0.5% for the rest of the tumor cell lines) for 72 hours in the presence of 5 μmol/L THC (A), different concentrations of THC ranging from 1 to 12 μmol/L (B), or the specified THC concentration (C). When antagonists were used (1 μmol/L rimonabant and 2 μmol/L SR144528), they were added 1 hour before THC. ER and p53 status was obtained from ATCC and German National Resource Centre for Biological Material (DSMZ, Braunschweig, Germany). IC50s were defined as concentrations of THC required to decrease cell viability to 50%. A, columns, mean for each cell line (n ≥ 3); bars, SE. B, significant differences from MCF-7 (*, P < 0.05; **, P < 0.01) or T-47D cells (‡, P < 0.05; ‡‡, P < 0.01). For real-time quantitative PCR experiments, an arbitrary value of 1 was assigned to cannabinoid receptor expression in MDA-MB 468 cells (reference). Representative experiment (n = 3). A and C, columns, means cell viability in THC-treated cells versus their respective vehicle-treated cells (set at 100%; n ≥ 3); bars, SE. Significant differences (*, P < 0.05; **, ††, P < 0.01) from control (*) or 5 μmol/L THC alone (†). D, CB1 and CB2 mRNA expression in the cell lines was determined by RT-PCR (representative gels, n ≥ 3). U373 MG astrocytoma cells and Jurkat leukemia cells were used as positive controls for CB1 and CB2, respectively.
THC-induced decrease of cell proliferation is due to the blockade of the G2-M transition. We next sought to examine whether an alteration of EVSA-T cell cycle underlies THC antiproliferative effect. The cannabinoid increased the number of cells in the G0-G1 compartment and, in parallel, decreased the number of cells in S phase ( Fig. 2A and C ). At the highest concentration tested (5 μmol/L), THC also produced the following: (a) an increase in the number of cells in G2-M phases and (b) the appearance of a population of hypodiploid cells ( Fig. 2A and C). The latter two effects were prevented by SR144528 ( Fig. 2C). Importantly, THC did not alter the cell cycle profile of HMEC cells ( Fig. 2B).
Figure 2.
Effect of THC on cell cycle dynamics. Cell cycle profiles of EVSA-T (A) and HMEC cells (B) incubated with 5 μmol/L THC or the corresponding vehicle for 48 hours. Representative experiments (n ≥ 3). C, percentage of EVSA-T cells (mean ± SE) in every phase of the cell cycle after 48 hours of incubation with the indicated compounds. Where indicated, rimonabant (SR1; 1 μmol/L) or SR144528 (SR2; 2 μmol/L) was added 1 hour before THC. Significant differences from control (*, P < 0.05; **, P < 0.01) or the corresponding concentration of THC alone (‡, P < 0.05).
To analyze the precise mechanism of THC action, we first studied the expression of several proteins involved in the G2-M transition. THC decreased the total levels of Cdc2 [p34, cyclin-dependent kinase (CDK) 1; Fig. 3A ], the major CDK controlling the entrance of cells in mitosis after completing G2 events ( 8), and SR144528 (2 μmol/L) completely prevented this effect [relative optical density (OD) after 16 hours of treatment relative to vehicle: SR144528+THC, 105 ± 4]. The expression of cyclin B1, the positive regulatory subunit of Cdc2 ( 9), did not significantly change on cannabinoid challenge (data not shown). The levels of p21, a CDK inhibitor known to prevent Cdc2-cyclin B activation ( 8), were enhanced by THC ( Fig. 3A). It has been recently proposed that p27, a CDK inhibitor traditionally associated to the regulation of G1-S transition, can also inhibit Cdc2 at G2-M (10). In our system, however, THC did not modify p27 levels (data not shown).
Figure 3.
Effect of THC on the expression of proteins involved in the control of the G2-M transition. A, Western blot analysis of the indicated proteins was done at least twice. Representative luminograms. T3, 3 μmol/L THC and T5, 5 μmol/L THC. Optical density (arbitrary units, mean ± SE) of the different proteins relative to their respective time-point control incubations (set at 100). B, EVSA-T cell viability was assayed after 72 hours of incubation with 5 μmol/L THC or vehicle. Columns, mean cell viability in THC-treated cells versus their respective vehicle-treated cells (set at 100%); bars, SE. Inset, Western blot of total Cdc2 levels in EVSA-T cells (lane 1), EVSA-T cells transfected with pIRESpuro2 (lane 2), and EVSA-T cells transfected with pIRESpuro2-Cdc2 (lane 3). A and B, significant differences from the corresponding vehicle-treated cells (*, P < 0.05; **, P < 0.01) or between THC-treated cells transfected with either pIRESpuro2 or pIRESpuro2-Cdc2 (‡, P < 0.05).
To be active, Cdc2 has to be dephosphorylated in the Tyr15 residue ( 11). THC treatment decreased the amount of phosphorylated Tyr15-Cdc2 to a lower extent than total Cdc2 levels ( Fig. 3A), indicating that the ratio inactive/active Cdc2 was augmented by THC. Phosphorylation of Cdc2 in Tyr15 is controlled by the Wee1/Mik1 family of protein kinases and by the phosphatase Cdc25C (11). Our results show that THC enhances Wee1 and reduces Cdc25C protein levels ( Fig. 3A).
In view of the aforementioned results, it is conceivable that THC exposure prevents EVSA-T cells to reach the required levels of active Cdc2 to enter mitosis. To test whether Cdc2 down-regulation is important in the growth-inhibiting effect of THC, we heterologously expressed this kinase. As shown in Fig. 3B, cells overexpressing Cdc2 became significantly more resistant to THC.
THC-induced cell cycle arrest is associated with apoptosis. We next tried to elucidate whether THC-induced inhibition of proliferation was associated with cell death. The cannabinoid induced apoptosis, a process that was prevented by SR144528 ( Fig. 4A ). THC also induced a two-fold increase in caspase-3 activity, an effect that was prevented by SR144528 ( Fig. 4B). Likewise, we observed reduced levels of both pro-caspase-3 (the inactive precursor of caspase-3) and PARP (a caspase-3 substrate) in cannabinoid-treated cells ( Fig. 4C). We subsequently addressed the question of whether apoptotic cells were those arrested in G2-M by THC. We conducted triple staining experiments to analyze the percentage of apoptotic cells in every phase of the cell cycle. As shown in Fig. 5A , THC induced apoptosis in all the cell cycle phases, but the majority of apoptotic cells were in the G2-M compartment.
Figure 4.
Analysis of THC-induced cell death. Fluorescence-activated cell sorting analysis (A) and caspase-3 activity (B) of EVSA-T cells after incubation with the indicated compounds for 24 hours. Antagonists (1 μmol/L rimonabant or 2 μmol/L SR144528) were added 1 hour before THC (5 μmol/L). A, percentage of apoptotic cells (FITC positive/PI negative) within the total cell population. B, columns, means of at least five or more experiments; bars, SE. Significant differences from control (*, P < 0.05; **, P < 0.01) or THC alone (‡, P < 0.05; ‡‡, P < 0.01). C, Western blot analysis of pro-caspase-3 and PARP. T3, 3 μmol/L THC and T5, 5 μmol/L THC. Representative luminograms (n ≥ 3).
Figure 5.
Connection between cell cycle arrest and apoptosis. A, percentage of apoptotic cells in different phases of the cell cycle as assessed by triple staining with Annexin V-FITC, PI, and Hoechst 33342. Significant differences from the corresponding vehicle-treated cells (*, P < 0.05; **, P < 0.01) or between THC-treated cells in G2-M and THC-treated cells in G0-G1 or S (‡, P< 0.05). B-D, Western blot of survivin in different human breast cell lines (B), EVSA-T cells incubated with THC (T3, 3 μmol/L THC; T5, 5 μmol/L THC) or the corresponding vehicle (C), and EVSA-T cells transfected with pIRESpuro2 or pIRESpuro-Cdc2 and incubated for 16 hours with the indicated compounds (D). Representative luminograms (n ≥ 2).
It is known that survivin, a member of the inhibitor of apoptosis family, can be phosphorylated in Thr34 by Cdc2. This phosphorylation results in enhanced stability of survivin and the consequent inhibition of caspase activity ( 12). Survivin was highly expressed in all the breast cancer cell lines tested but was hardly detectable in HMEC ( Fig. 5B), in agreement with previous reports showing a sharp differential expression in cancer (high levels) versus normal (undetectable levels) tissues (12). Moreover, THC decreased survivin levels in EVSA-T cells ( Fig. 5C), which may explain why THC-induced Cdc2 inactivation results in apoptosis. In fact, when Cdc2 levels were enhanced by overexpression, survivin decrease on cannabinoid treatment was attenuated (relative optical density after 16 hours of treatment for pIRESpuro2-transfected cells: vehicle, 100 ± 6; 5 μmol/L THC, 31 ± 4. For pIRESpuro2-Cdc2-transfected cells: vehicle, 100 ± 5; 5 μmol/L THC, 74 ± 3; Fig. 5D).
Cannabinoid receptors are expressed in human breast tumors. The presence of CB1 and CB2receptors in human breast tumors was evaluated by real-time quantitative PCR and confocal microscopy. Lower levels of CB1 mRNA were detected in tumors of low-medium (grade 1-2) and high (grade 3) histologic grade compared with normal, noncancerous breast tissue (grade 1-2 versus noncancerous breast tissue (P = 0.008); grade 3 versus noncancerous breast tissue (P = 0.0007); Fig. 6A ). CB2 expression was higher than CB1 expression in all the tumors analyzed (P = 0.00002) and seemed to correlate with their histologic grade (grade 1-2 versus grade 3; P = 0.04; Fig. 6A). Of interest, CB2 transcripts were hardly detectable in normal breast tissue ( Fig. 6A). Moreover, the expression of CB2 showed an association with molecular markers of prognostic value. Thus, ER− tumors expressed more CB2 mRNA than ER+ tumors ( Fig. 6B). CB2 expression was also higher in PR− than in PR+ samples. ER−/PR− tumors (response rate <10% to conventional therapies) expressed more CB2 mRNA than ER+/PR+ tumors (60-70% response rate; ref. 7; Fig. 6B). ERBB2/HER-2-positive tumors (with worse survival prediction at least in node-positive patients; ref. 7) expressed higher levels of CB2 mRNA than ERBB2/HER-2-negative tumors ( Fig. 6B). Confocal microscopy experiments confirmed the presence of CB1 and CB2 proteins in tumor cells ( Fig. 6C). None of the receptor proteins was detected in normal ducts.
- Figure 6.
Cannabinoid receptor expression in human breast tumors. A and B, mRNA expression (medians ± SE) of CB1 and CB2receptors as determined by real-time quantitative PCR. An arbitrary value of 1 unit was assigned to CB2 expression in one of the tumors (reference), and the relative expression of the rest of the samples was calculated by comparison with the reference. Note the different scale in the y axes of (A). Significantly different values (*, P < 0.05; **, P < 0.01) from “+ variable” or “+/+ variable.” ER; PR; S1, <2 cm; S2, 2 to 5 cm; and S3, >5 cm; metastasis was determined as presence/absence of positive axillary lymph nodes. C, confocal microscopy analysis of cannabinoid receptors in a grade 1 (left) and a grade 3 tumor (right). Green, cell nuclei; red, cannabinoid receptors. Arrows, different areas within the tumor samples.
There is scant information on the effect of cannabinoids on the cell cycle. It has been shown previously that the endogenous cannabinoid anandamide arrests the cycle of hepatoma HepG2 cells ( 13), epidermal growth factor–stimulated PC3 prostate cancer cells ( 14), and the breast cancer cell line EFM-19 ( 15) at the G1-S transition. The same effect was exerted by a metabolically stable analogue of anandamide, met-F-anandamide, on KiMol thyroid carcinoma cells ( 16). Results presented herein (a) show that the plant-derived cannabinoid THC is able to block the progression of breast cancer cell cycle and (b) provide a mechanism for this action. Of interest, nontumor mammary epithelial cells were rather insensitive to THC, suggesting that cannabinoids would fulfill one of the demanded requirements of any compound intended to be used in cancer therapy: the selectivity for tumor cells. In our experimental model, THC induces a CB2-mediated cell cycle arrest at the G2-M transition via Cdc2 down-regulation. We are nonetheless aware that our observation that THC effects are significantly but not completely prevented by the CB2 selective antagonist also points to the existence of CB2-independent processes in cannabinoid antiproliferative action. Most cancer cells evade antigrowth signals because they have defective G1 checkpoints, which makes the G2 checkpoint an attractive target for cancer therapy ( 11). Since its discovery in 1986 ( 17), it has been clear that Cdc2 is essential for cell cycle progression ( 18). In fact, Cdc2-deficient mice die at very early stages of embryonic development ( 18). The pivotal importance of this particular protein could explain why, in our system, a somewhat moderate decrease of total Cdc2 levels (∼40%) results in large changes in cell viability. In addition, our data indicate that THC increases the inactive/active Cdc2 ratio, supporting that the cannabinoid decreases not only total Cdc2 levels but also enzyme-specific activity.
It is important to point out that, although the antiproliferative effect of cannabinoids on different tumor cells has been extensively confirmed both in vitro and in vivo ( 6), a recent report indicates that THC may enhance breast cancer cell growth under certain circumstances. In that study, the authors showed a direct association between the degree of sensitivity of a tumor to THC and the level of CB1 and CB2 expression. Thus, THC has antiproliferative effect in tumors expressing cannabinoid receptors, whereas those with low to no expression suffer increased growth and metastasis due to THC-induced suppression of the antitumor immune response ( 19). Results presented herein are not in disagreement with that report, as EVSA-T cells, which are very sensitive to THC, express high levels of CB2.
By real-time quantitative PCR experiments, we have observed a correlation between CB2expression and the histologic grade of breast tumors. Moreover, CB2 expression is higher in tumors with predicted low response to conventional therapies, for instance ER−/PR− tumors, which are weakly responsive to adjuvant tamoxifen ( 7). In addition, hardly detectable levels of CB2 were found in normal breast tissue. Because both cell cycle arrest and apoptosis induced by THC are CB2-mediated effects, it is tempting to speculate that the tumors with poor prognosis (i.e., those reluctant to conventional therapies and, according to our data, expressing the highest levels of CB2receptor), may be the most responsive to cannabinoids. In addition, the psychotropic effects of cannabinoids are mediated by CB1 but not CB2 receptors, and, therefore, a cannabinoid-based therapy selectively targeting CB2 receptors would be devoid of the side effects associated to cannabis consumption ( 20).
Breast cancer is the most common malignant disease among Western women. Although the rates of mortality of breast cancer patients have decreased as a result of early diagnosis by mammograms, certain breast tumors remain reluctant to conventional therapies, and current treatments have side effects that substantially affect the patient's quality of life ( 21). Our findings might set the basis for new strategies for the management of breast cancer.
Acknowledgments
Grant support: Fondo de Investigaciones Sanitarias FIS 04/1029 (C. Sánchez) and BEFI 01/9132 (D. Sarrió); Complutense University PR1/05 (C. Sánchez); Ministerio de Educación y Ciencia SAF2003-00745 (M. Guzmán), SAF2004-08258-C02-01 (J. Palacios), and AP2003-0063 (M.M. Caffarel); and Fundación Científica de la Asociación Española Contra el Cáncer (M. Guzmán).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank the staff at Centro de Microscopía y Citometría (Complutense University, Madrid, Spain) and A. Álvarez-Barrientos for expert advice on flow cytometry experiments; O. Rueda for expert advice on statistical analysis; and the members of our laboratory for technical support, especially to K. Blazej, C. Blázquez, D. Callegarin, A. Egia, B. Herrera, and M. Lorente.
Footnotes
- Received December 22, 2005.
- Revision received April 6, 2006.
- Accepted May 2, 2006.
- ©2006 American Association for Cancer Research.
- ↵
Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001;1: 222–31.
CrossRefPubMed - ↵
Guzman M, Sanchez C, Galve-Roperh I. Cannabinoids and cell fate. Pharmacol Ther 2002; 95: 175–84.
CrossRefPubMed - ↵
Velasco G, Galve-Roperh I, Sanchez C, Blazquez C, Guzman M. Hypothesis: cannabinoid therapy for the treatment of gliomas? Neuropharmacology 2004; 47: 315–23.
CrossRefPubMed - ↵
Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc 1964; 86: 1646–47.
CrossRef - ↵
Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54: 161–202.
Abstract/FREE Full Text - ↵
Guzman M. Cannabinoids: potential anticancer agents. Nat Rev Cancer 2003; 3: 745–55.
CrossRefPubMed - ↵
Tavassoli F, Devilee P, editors. World Health Organization: tumours of the breast and female genital organs (IARC/World Health Organization Classification of Tumours). Lyon: IARC Press-WHO; 2003.
- ↵
Stark GR, Taylor WR. Analyzing the G2-M checkpoint. Methods Mol Biol 2004; 280: 51–82.
PubMed - ↵
Smits VA, Medema RH. Checking out the G2/M transition. Biochim Biophys Acta 2001; 1519: 1–12.
PubMed - ↵
Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6: 661–72.
CrossRefPubMed - ↵
Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004; 3: 513–9.
Abstract/FREE Full Text - ↵
Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003; 22:8581–9.
CrossRefPubMed - ↵
Biswas KK, Sarker KP, Abeyama K, et al. Membrane cholesterol but not putative receptors mediates anandamide-induced hepatocyte apoptosis. Hepatology 2003; 38: 1167–77.
CrossRefPubMed - ↵
Mimeault M, Pommery N, Wattez N, Bailly C, Henichart JP. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 2003; 56: 1–12.
CrossRefPubMed - ↵
De Petrocellis L, Melck D, Palmisano A, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci U S A 1998; 95: 8375–80.
Abstract/FREE Full Text - ↵
Portella G, Laezza C, Laccetti P, De Petrocellis L, Di Marzo V, Bifulco M. Inhibitory effects of cannabinoid CB1 receptor stimulation on tumor growth and metastatic spreading: actions on signals involved in angiogenesis and metastasis. FASEB J 2003; 17: 1771–3.
Abstract/FREE Full Text - ↵
Russell P, Nurse P. Schizosaccharomyces pombe and Saccharomyces cerevisiae: a look at yeasts divided. Cell 1986; 45: 781–82.
CrossRefPubMed - ↵
Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005; 30: 630–41.
CrossRefPubMed - ↵
McKallip RJ, Nagarkatti M, Nagarkatti PS. Δ-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol 2005; 174: 3281–9.
Abstract/FREE Full Text - ↵
Sanchez C, de Ceballos ML, del Pulgar TG, et al. Inhibition of glioma growth in vivo by selective activation of the CB2 cannabinoid receptor. Cancer Res 2001; 61: 5784–9.
Abstract/FREE Full Text - ↵
Edwards BK, Brown ML, Wingo PA, et al. Annual report to the nation on the status of cancer, 1975-2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst 2005; 97: 1407–27.
Abstract/FREE Full Text